Company Intelligence Brief

What deterministic perturbation analysis suggests about distributed biological architecture
Matthew L. Hardy | NomosLogic Inc.
Human genetics has become extraordinarily good at finding associated variants. It has been less successful at answering a harder question: what kind of biological system produces those associations in the first place?
For years, much of the field has worked inside an implicit model. Complex disease is often treated as the sum of variant effects, with the most statistically prominent loci frequently interpreted as the most important biological contributors. That framing has been productive, but it may also be incomplete.
At NomosLogic, we have been studying a different possibility: that many biological systems are organized not around a handful of structurally essential variants, but around distributed constraint architectures, interacting sets of variants that preserve stable system behavior even when apparently dominant contributors are removed.
That distinction matters because it changes the question. Instead of asking only which variant is most prominent, it asks what kind of system can preserve stable behavior under perturbation.
What we observed
Using PROTEUS, our deterministic evolutionary simulation framework, we tested whether genomic systems collapse under targeted perturbation or reorganize while preserving convergence.
The results were striking.
Across the observed dataset, 88.5% of systems exhibited distributed architecture. Across perturbation experiments, no exclusion caused loss of convergence. In a cardiovascular test case, removing the dominant contributor preserved 99.996% of peak fitness, accelerated convergence from generation 140 to generation 90, and produced 29 novel variants rising into top-ranked positions. Validation against empirical null distributions yielded AUC 0.9386, indicating that the framework was identifying biological signal rather than optimization artifact.
That pattern is difficult to reconcile with a simple variant-centric model of causality. It is much easier to reconcile with the idea that stable biological behavior emerges from distributed interacting architectures that can reorganize under pressure while preserving system-level structure.
Summary of findings:
88.5% distributed architecture
no perturbation caused loss of convergence
99.996% fitness retention
29 novel variants rose after exclusion
AUC 0.9386
Why determinism matters
This is also why determinism matters.
Many computational approaches in genomics are stochastic. That is acceptable for some questions, but it makes perturbation interpretation harder. If identical inputs do not reliably produce identical outputs, then it becomes more difficult to distinguish biological structure from computational noise.
PROTEUS was designed to be deterministic. Identical inputs produce identical outputs. That design choice was not cosmetic. It is what made rigorous perturbation analysis possible. The manuscript defines convergence in deterministic terms and states explicitly that all simulations are deterministic across independent runs with identical inputs.
The goal was not simply to optimize a score. It was to test whether convergence is an intrinsic property of the system itself.
The evidence suggests that it is.
Why the field should care
If this view is right, the consequences extend beyond theory.
Variant interpretation, therapeutic targeting, and risk modeling all depend on how causality is framed. If the field overweights the apparent dominance of individual contributors, it may end up targeting concentration points rather than structural requirements.
That does not make current approaches useless. Polygenic methods, association statistics, and fine-mapping remain valuable. But they may be incomplete if they assume that the most visible contributors are also the most architecturally necessary.
A system can produce a strong signal around a variant without being fundamentally dependent on that variant for stable behavior.
That distinction matters for drug development as much as for interpretation. A target may be prominent, measurable, and clinically associated, yet still not be the right abstraction for understanding how a biological system maintains itself. The manuscript states this directly, noting that therapeutic strategies aimed at single “driver” variants may be addressing concentration points rather than causal requirements.
A more useful question
The field may now need to ask a different question.
Instead of asking only which variant is most important, genomics may need to ask what kind of system preserves stable behavior even when its most visible contributors are removed.
That shift sounds subtle, but it is foundational.
It moves the problem from ranking contributors to understanding architecture. It also offers a more biologically plausible way to think about complex disease, not as a brittle stack of independent effects, but as a constrained adaptive system whose behavior is distributed across interacting components.
What this could change
If distributed constraint architecture is real, and the perturbation results strongly suggest that it is, then some of the most important work ahead will not be about finding ever more significant variants.
It will be about identifying how biological systems remain coherent, how they reorganize under perturbation, and how that organization can be measured without confusing prominence for necessity.
Genomics does not need less statistical power.
It may need a broader model of causality.
Bottom line
The central claim is straightforward:
In many genomic systems, causality may be distributed across interacting variant sets rather than concentrated in a few dominant anchors.
If that is true, then the field has an opportunity to move beyond a ranking logic that has become familiar and toward an architectural logic that may be closer to how biology actually works.
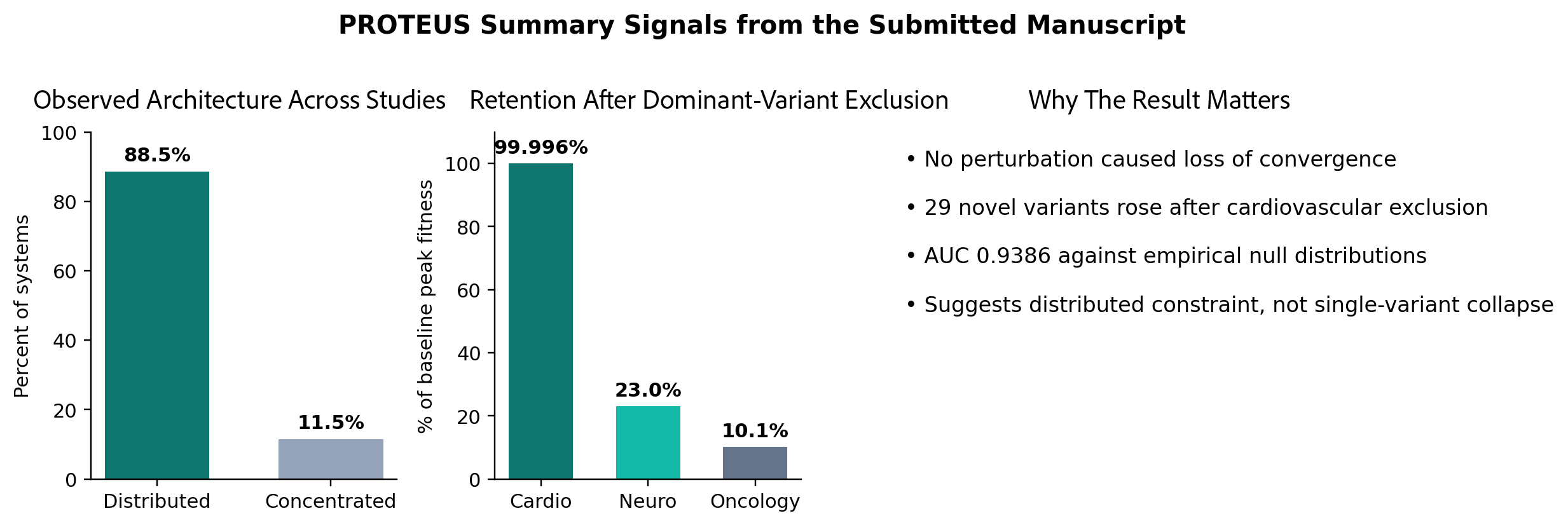
Figure. Summary signals drawn from the submitted manuscript: most observed systems exhibited distributed architecture; convergence persisted under dominant-variant exclusion; and validation against null distributions was strong.