The prevailing computational model in human genetics assumes that variants contribute additively and, to a first approximation, independently to phenotype. Polygenic risk frameworks extend this logic by aggregating many small effects into a composite score. Although valuable in many contexts, these approaches do not fully reflect how biological systems function.
Biology is not additive by construction. Proteins interact, pathways constrain one another, regulatory programs are layered, and cellular stress is often mediated through coupled systems rather than isolated loci. If this is true at the biological level, then genetic interpretation may require models capable of identifying structured interaction rather than simply accumulating effects.
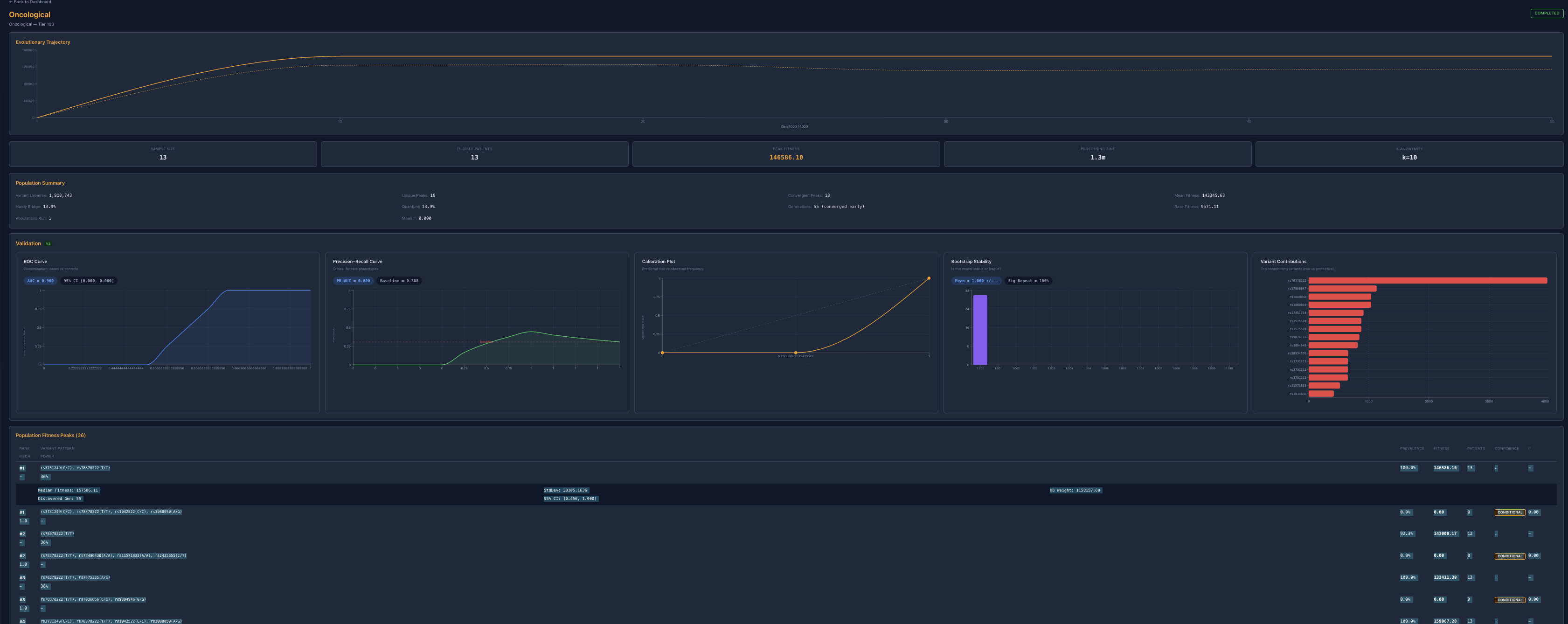
Proteus V3 was developed to explore that possibility. Rather than ranking variants in isolation, the framework searches for multi-variant configurations and subjects them to repeated validation pressure, including cross-validation, permutation-based null modeling, and bootstrap stability analysis. The goal is not merely to maximize association, but to retain only those combinations that remain stable, reproducible, and biologically interpretable.
Across cardiovascular, oncological, metabolic, and neurological analyses, a consistent pattern emerged: complex genomic space often resolves into low-dimensional hierarchy.
In some domains, that hierarchy appears reducible. Cardiovascular analyses repeatedly converged on a dominant structural axis with a smaller metabolic modifier layer. Oncological analyses similarly converged on a primary regulatory anchor, with secondary variants contributing little incremental explanatory value. These systems appear compressible: although many variants are present, only a small number define the core structure of the signal.
In other domains, however, the system does not reduce to a single anchor. Neurological analyses suggest a different form of organization, in which multiple variants corresponding to distinct biological functions persist together as a stable cluster. Rather than collapsing to one dominant driver, the solution appears to require simultaneous integrity across protein handling, transcriptional regulation, and degradation-related pathways. This is consistent with the underlying biology of neuronal systems, where distributed dependencies are expected.
These observations suggest that not all phenotypes are organized in the same way. Some may be governed primarily by a dominant genetic axis, whereas others depend on co-equal, interacting constraints. This distinction introduces a more nuanced view of genetic architecture than either single-variant association or diffuse polygenic accumulation.
A second notable observation is the systematic rejection of many conditional variant combinations. Across domains, high-complexity combinations frequently failed to persist under validation, often showing zero prevalence or no reproducible contribution to fitness. In effect, the framework filtered much of the apparent combinatorial complexity out of the system. This suggests that a substantial portion of high-dimensional genomic space may represent unstable or non-informative structure rather than durable biological signal.
Importantly, these observations should be interpreted conservatively. Current analyses remain limited by sample size, cohort composition, and incomplete population diversity. The results do not establish causality, nor do they yet justify broad clinical claims. What they do provide is a repeatable computational observation: when subjected to iterative validation, genomic systems may reveal hierarchical structure rather than diffuse additive contribution.
If validated in larger and more heterogeneous cohorts, this would have meaningful implications. It would suggest that genetic interpretation should move beyond the assumption of independent, additive effects and toward a framework that asks a different question: not simply how much risk is present, but how the system itself is organized.
That shift matters because it makes interpretation more biologically legible. A hierarchy can distinguish dominant from secondary contributors. A distributed cluster can reveal when simplification fails. A rejected conditional pattern can indicate where complexity is only noise. These are not just technical improvements; they are conceptual ones.
The next phase is clear. The hypotheses generated by these interaction hierarchies must be challenged across larger cohorts, diverse populations, and external validation settings. If they persist, then genomic modeling may need to be understood less as a problem of aggregation and more as a problem of structure.
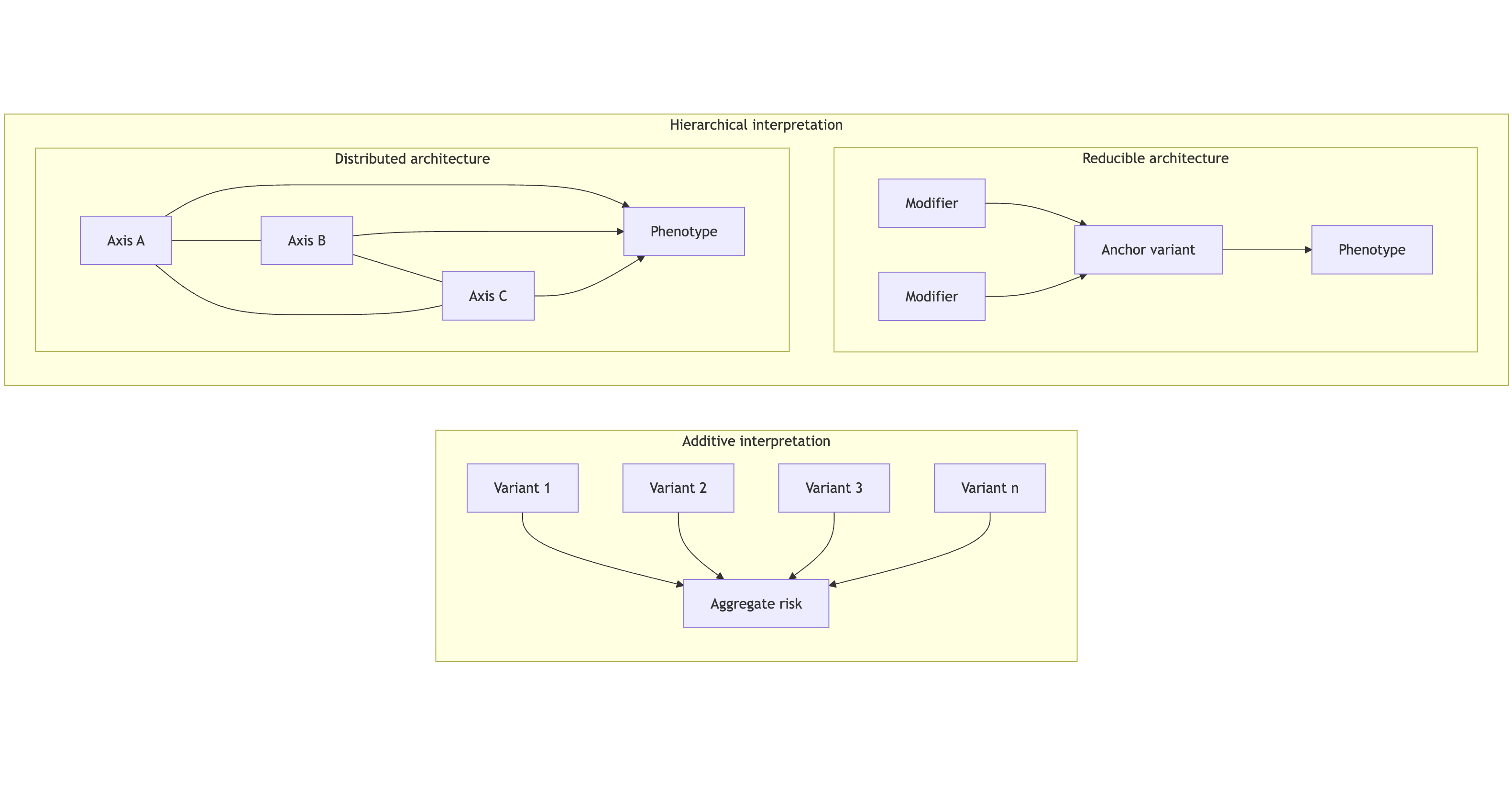
Figure 1 caption: Additive models assume independent variant effects that sum into a downstream estimate of risk. A hierarchical interpretation instead distinguishes between reducible systems, which collapse to a dominant axis, and distributed systems, which require multiple co-dependent axes to explain phenotype expression.
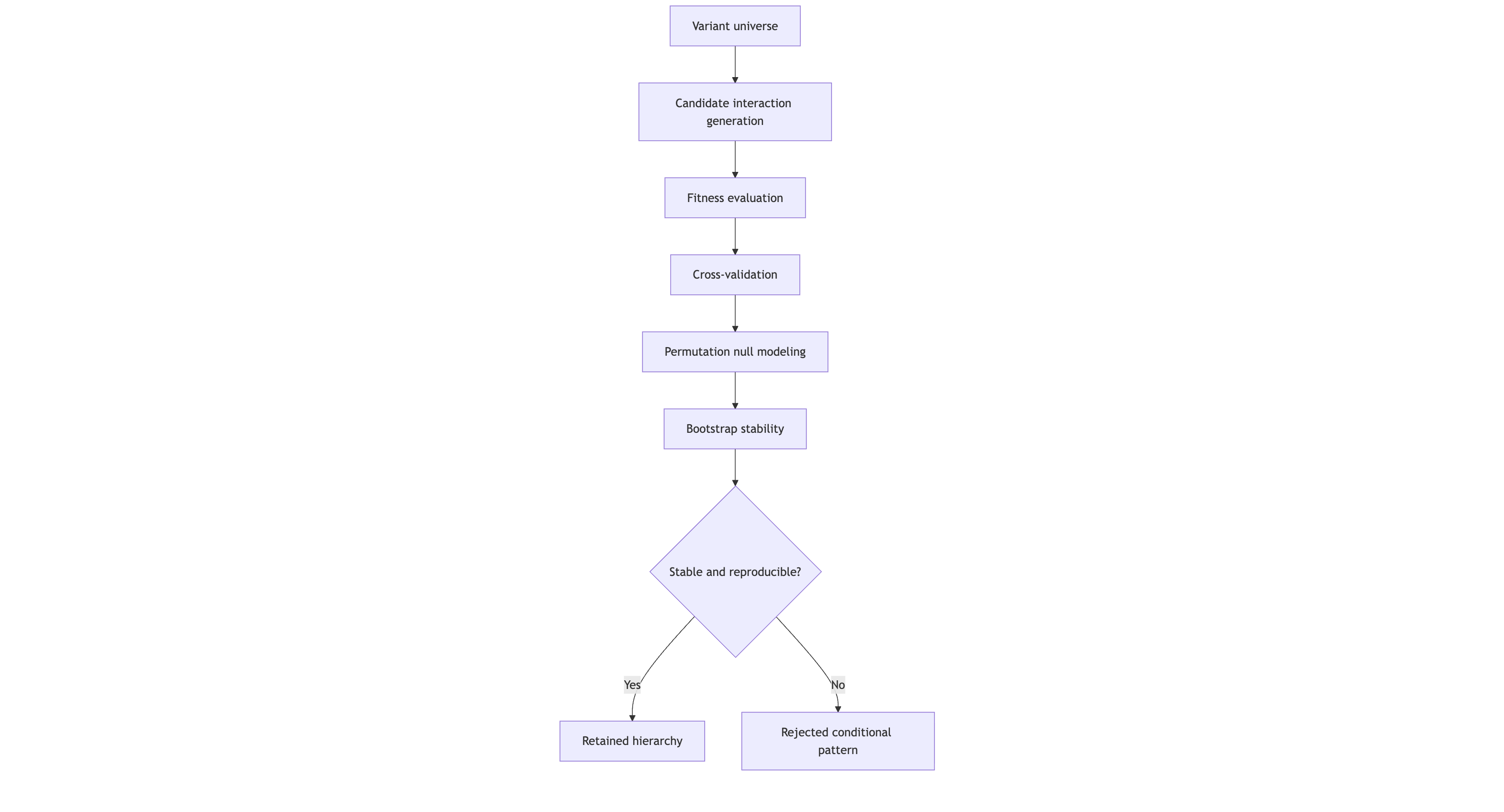
Figure 2 caption: Proteus V3 evaluates candidate multi-variant configurations through repeated validation pressure. Only those structures that remain stable under cross-validation, null perturbation, and bootstrap resampling are retained as interpretable hierarchies; unstable combinations are rejected.