Article
Modern genomics has a signal problem.
Sequencing has made it possible to observe millions of variants across individuals, but interpretation still lags behind data generation. Most computational approaches remain anchored to additive models, where variants contribute independently to a downstream estimate of risk. Those models can be useful, but they often trade away interpretability and struggle to distinguish meaningful structure from combinatorial noise.
The result is a familiar paradox: more data, but not necessarily more understanding.
An alternative approach is to model genetic systems as structured rather than additive.
Instead of asking which variants contribute independently to phenotype, a systems-based framework asks whether variants organize into stable, interacting structures. In practice, that means treating the genome less like a list of independent inputs and more like a constrained system with dominant axes, modifiers, and failure points.
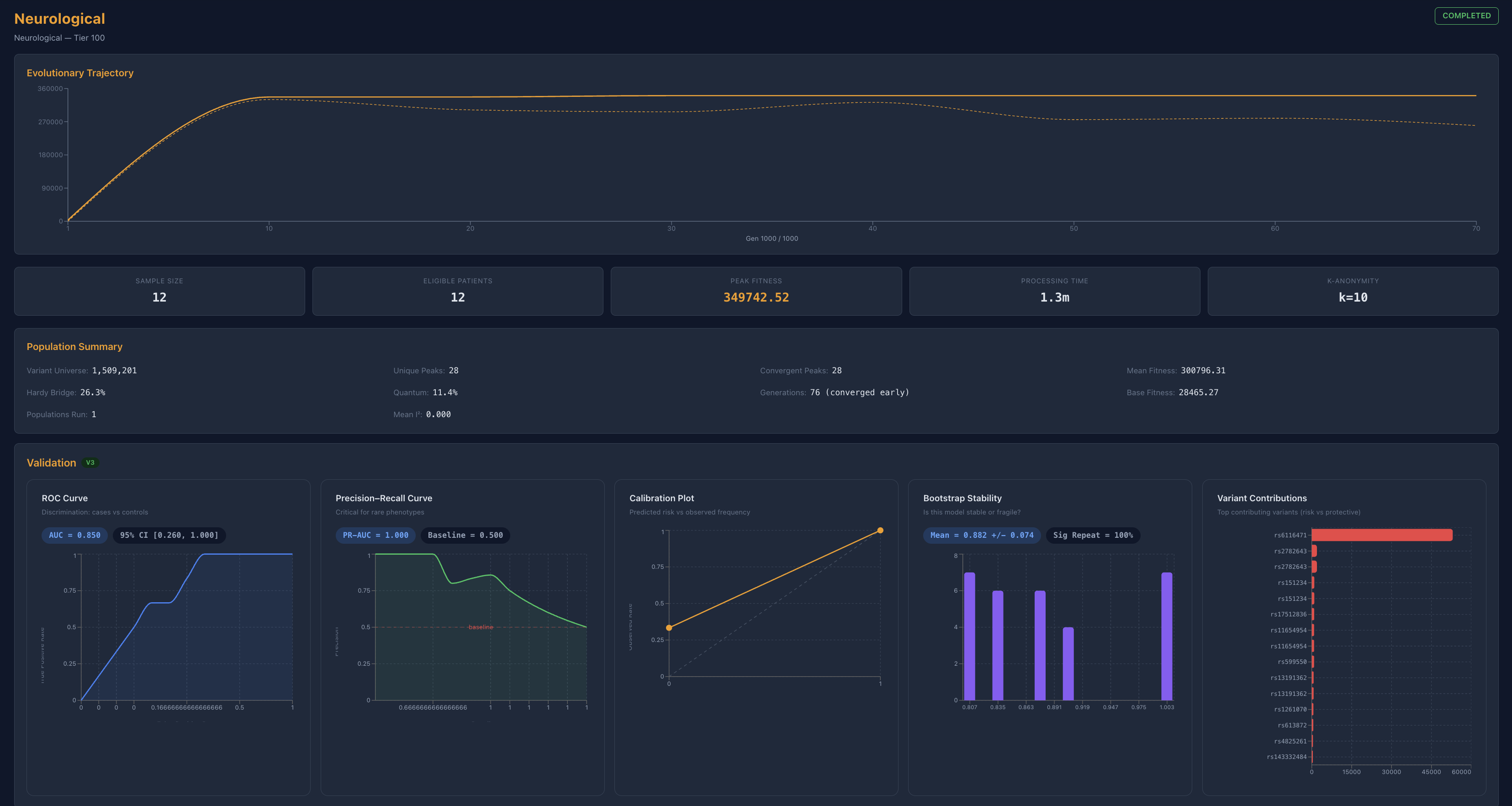
That is the premise behind Proteus V3, an evolutionary optimization framework designed to identify multi-variant interaction structures under validation pressure. Candidate configurations are evaluated not only for fitness, but also for reproducibility across cross-validation, permutation-based null modeling, and bootstrap resampling. Biological plausibility can be incorporated through pathway and network constraints, allowing the system to prioritize structured signal over merely frequent co-occurrence.
Across cardiovascular, oncological, metabolic, and neurological analyses, one pattern repeatedly emerges: high-dimensional genomic space often compresses into low-dimensional, interpretable structure.
In some domains, the system collapses to a dominant axis. In cardiovascular analyses, for example, structural cardiac signal may dominate, with metabolic variants acting as secondary modifiers. In oncological analyses, a master regulatory locus may anchor the solution space, while additional variants contribute only marginally. In these cases, the system appears reducible: most of the explanatory weight can be captured by a small number of variants.
In other domains, especially those associated with distributed biological function, the system does not collapse to a single dominant signal. Instead, multiple co-dependent axes persist together. Neurological analyses suggest exactly this kind of structure, where protein integrity, transcriptional regulation, and clearance-related pathways appear as a stable cluster rather than a single anchor. In these cases, the system behaves less like a line and more like a network.
This distinction matters. If some biological systems are reducible while others require network integrity, then additive approaches are not just incomplete; they are blind to the organizational form of the underlying biology.
The computational implications are equally important. When genomic systems are modeled hierarchically, dimensionality reduction becomes structural rather than purely statistical. Instead of collapsing data into an opaque risk score, the model can identify what matters most, what modifies it, and which combinations fail to reproduce under validation. That makes the output more interpretable and potentially more actionable.
It also changes the architecture of decision systems. In a conventional pipeline, discovery, interpretation, and application are often disconnected. A signal may be found in one environment, manually translated in another, and only weakly connected to downstream decision support. In a systems-oriented architecture, discovery can be embedded within a controlled environment, translated through deterministic logic, and surfaced through application interfaces without breaking the chain between data, structure, and decision.
That does not eliminate the need for caution. These findings remain early and require broader validation, especially across larger and more diverse cohorts. Small sample sizes, shared baseline characteristics, and domain-specific cohort effects can all distort apparent structure. The key question is not whether every emerging hierarchy is true, but whether the same organizational patterns persist under repeated attempts to falsify them.
That is the real opportunity. If genetic systems are architectural rather than merely additive, then the next era of genomics may depend less on collecting ever-larger variant catalogs and more on learning how to detect the structure already present within them.
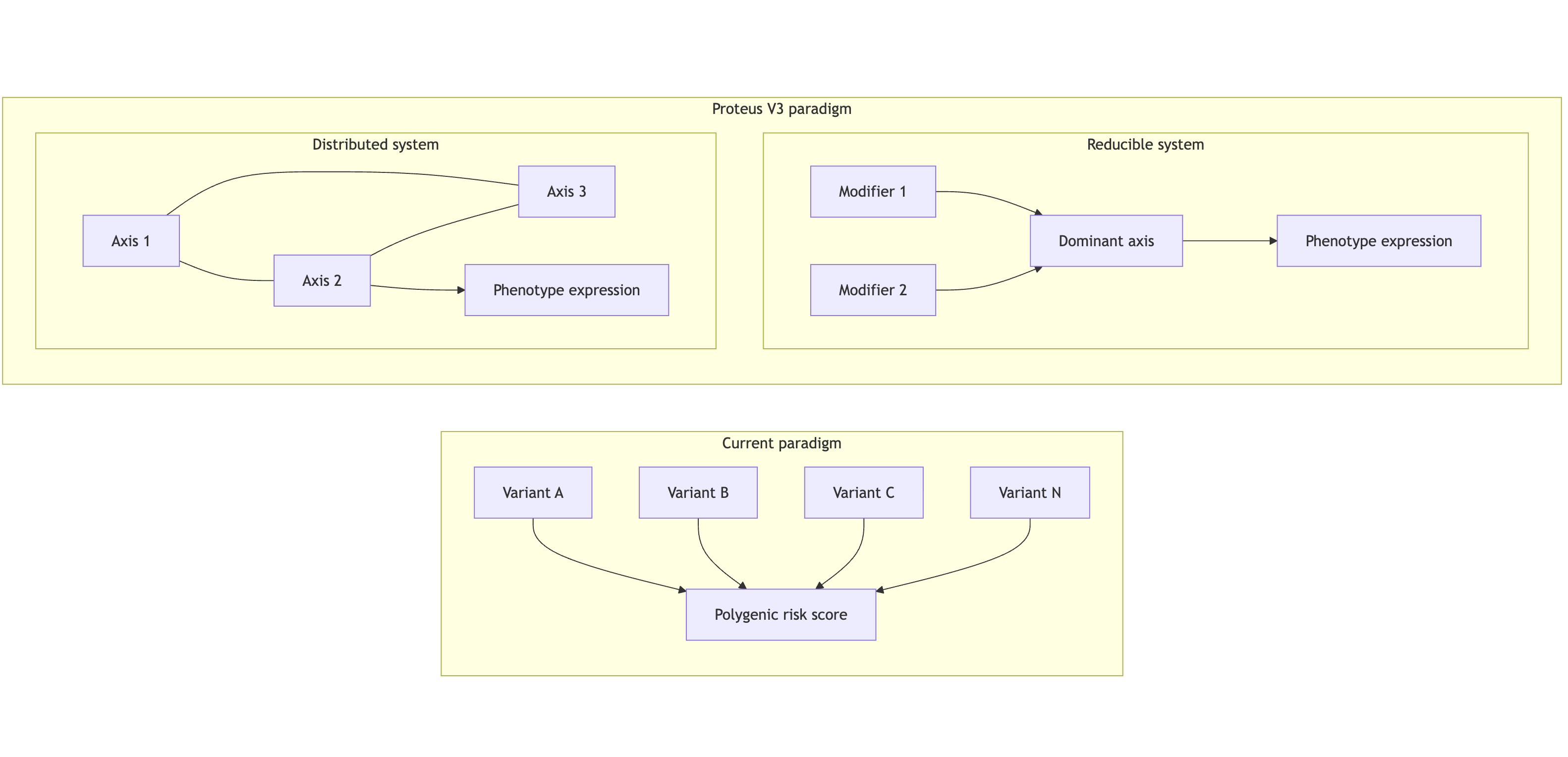
Figure 1 — Paradigm shift
Figure 1 caption: Traditional genomic models treat variants as independent contributors to downstream risk. In contrast, a systems-based approach identifies hierarchical organization in genetic space. Some domains collapse to a dominant axis with secondary modifiers, while others require coordinated integrity across multiple co-dependent axes.