The Architecture is Reading Two Diseases Differently. That is the Point.
Two cohort studies. Same framework. Two architecturally distinguishable empirical signatures. What distributed constraint architecture actually looks like when you read it directly.
Matthew L. Hardy | Founder & CEO, NomosLogic
The platform finished two cohort studies this week. A diabetes cohort and a hematology cohort. Same simulation framework, same disclosure architecture, same validator. Two completely different empirical signals.
That difference is the finding.
Diabetes: the architecture reorganized rather than collapsed.
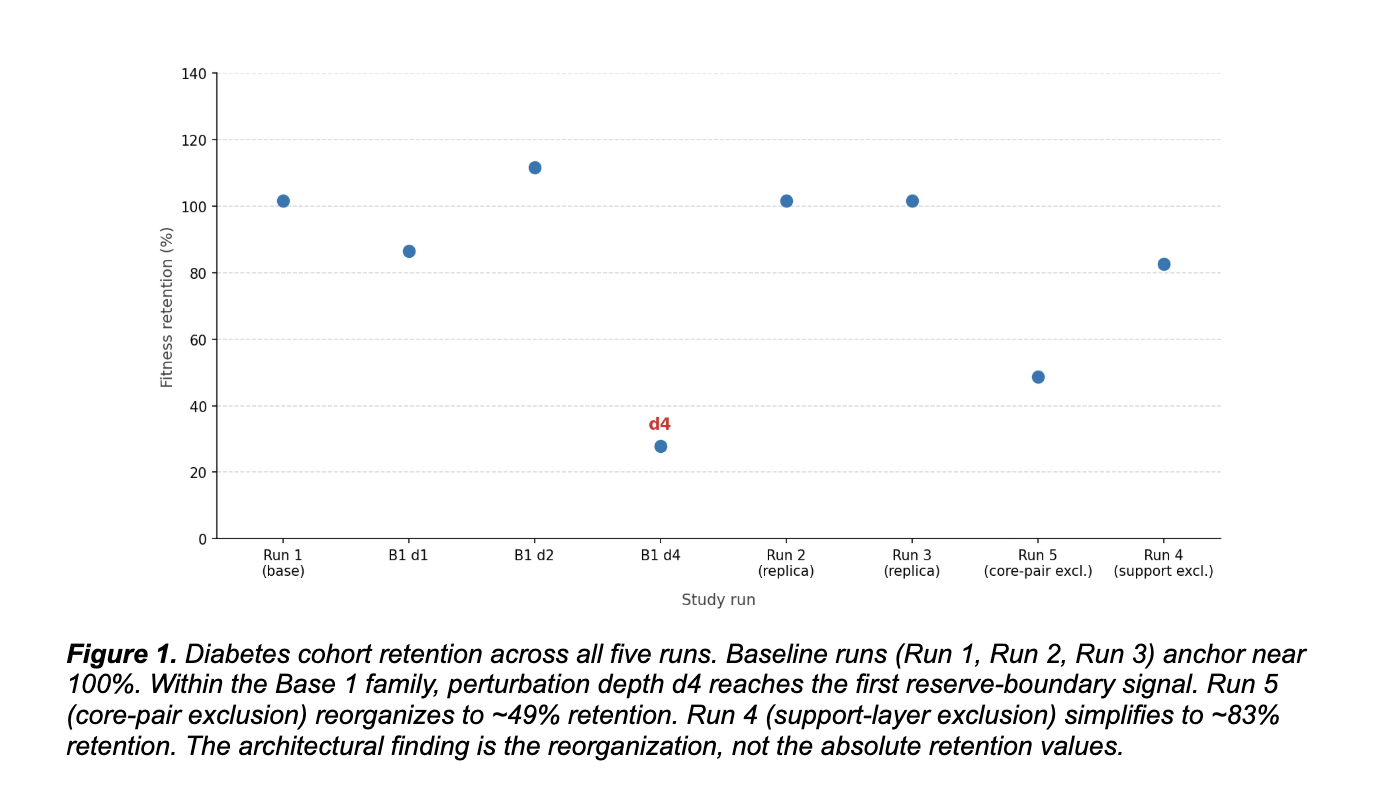
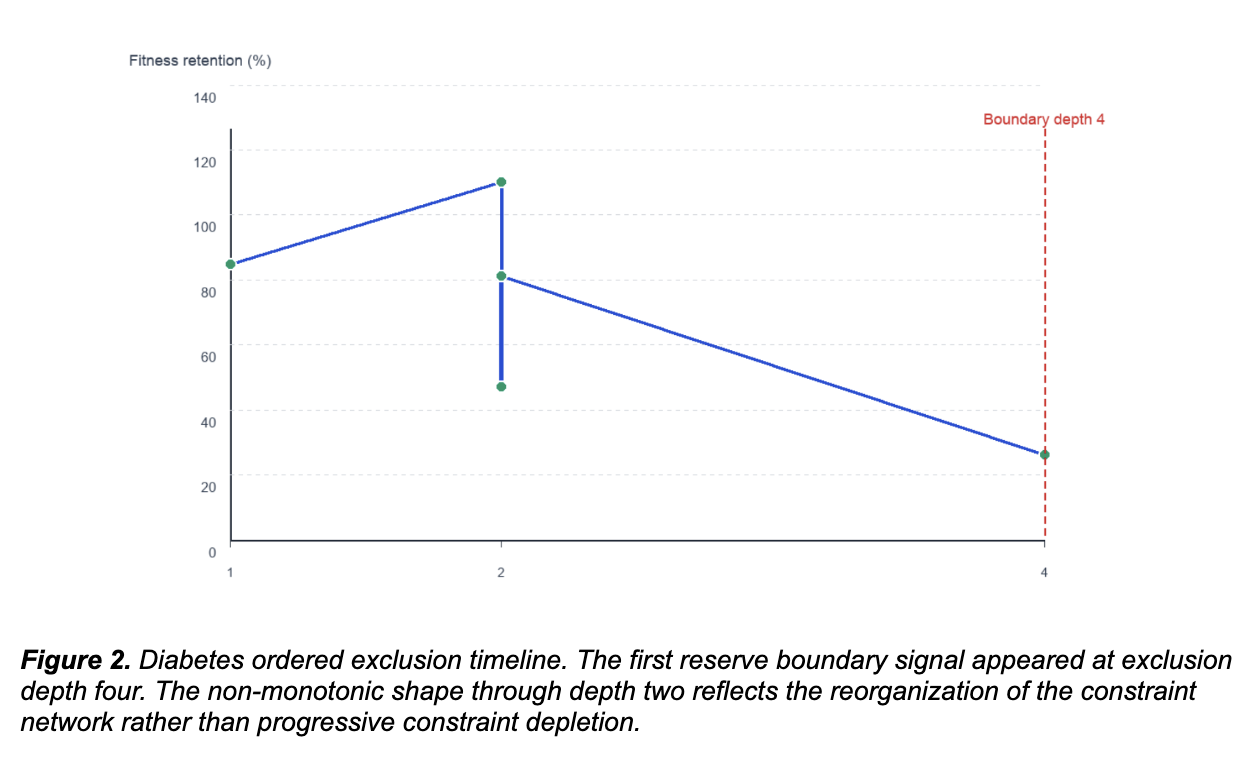
In the diabetes cohort, the system recovered a stable variant family across replicate runs under fixed conditions. Run 1 established a compact base family. Runs 2 and 3 reproduced that core and consolidated it through replication. The same family returned under identical settings rather than drifting to unrelated peaks. That kind of recurrence under matched conditions is what reproducibility looks like in a constraint-architecture framework.
Then we excluded the dominant pair. Run 5 removed the two variants that anchored the base family. Under standard pharmacogenomic logic, the system should have lost coherence. It did not. It reorganized into a second structured family centered on different variants, with top patterns repeatedly involving the same components at 80% prevalence, convergence remaining early, and bootstrap repeatability holding strong. AUC stayed at 1.000.
The system did not collapse because the constraint architecture had redundancy. The dominant pair was not the only configuration the network supported. There was an alternative coherent solution available, and the system found it. That is reconfiguration, not failure.
Hematology: the architecture depleted progressively rather than reorganized.
In the hematology cohort, we ran progressive multi-variant exclusion through depth seven. Retention declined approximately monotonically from 98% at depth one to 15% at depth seven. No alternative coherent family emerged. The system did not find a second attractor. It depleted constraint until reaching a finite reserve boundary.
Validation metrics remained strong throughout the series. Bootstrap stability mean ranged from 0.805 to 1.000. Top-signature repeatability ranged from 80% to 100%. The progressive decline was reproducible, not stochastic. The architecture has less redundancy than the diabetes cohort. Progressive exclusion depleted the reserve because the constraint network could not redistribute beyond a defined depth.
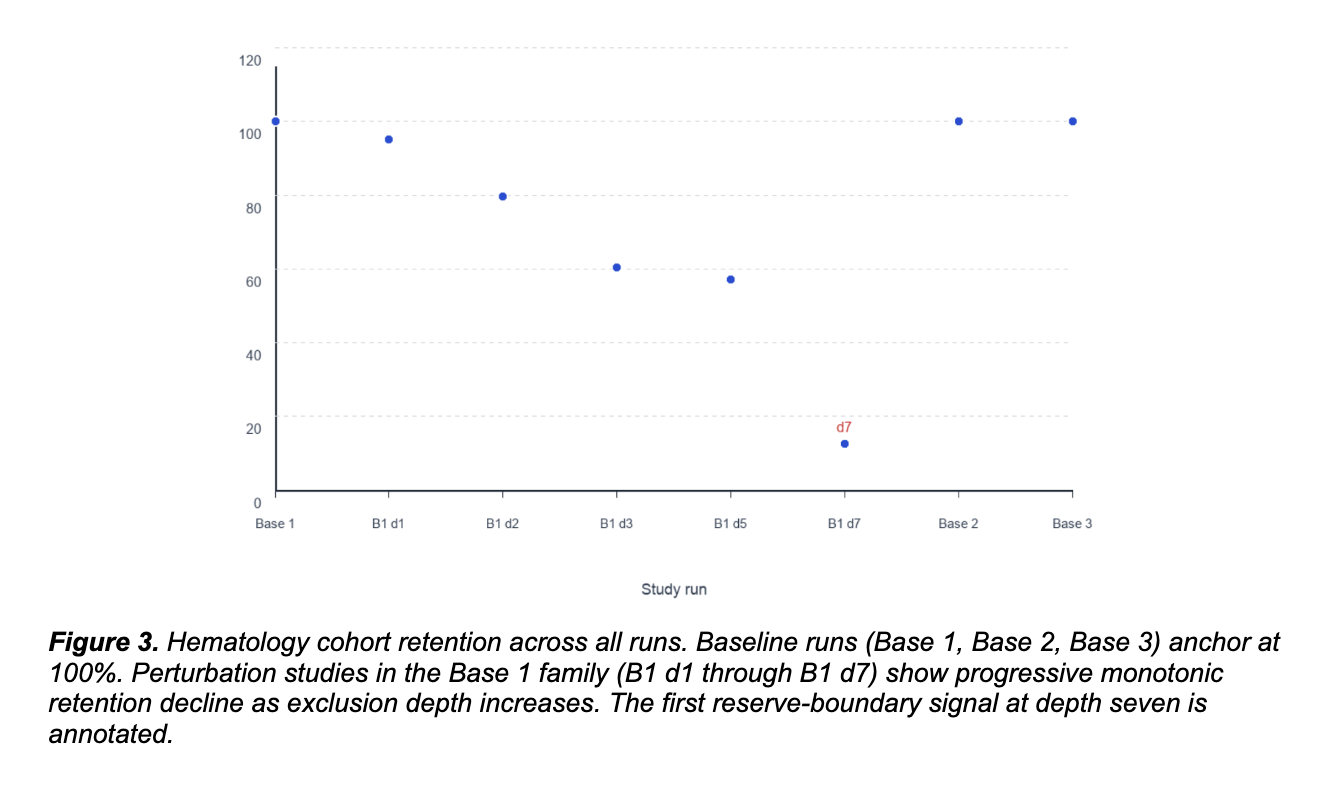
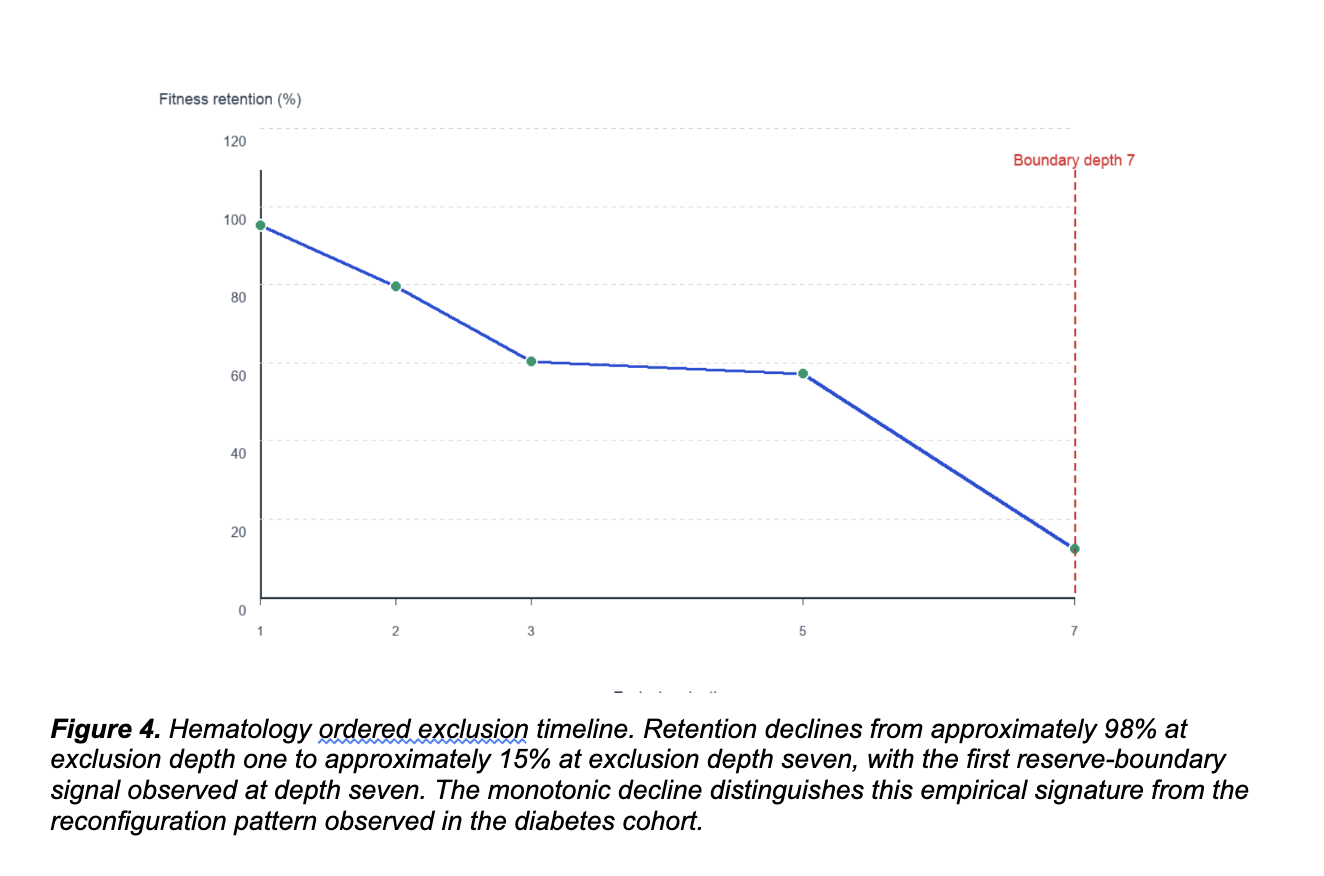
Two empirical signatures. One framework. The point is the difference.
Distributed constraint architecture is not a uniform property across clinical domains. It varies. A framework that reads architecture correctly produces different empirical signals for different domains, because the underlying biological architectures are different.
Population-averaged variant scoring cannot produce these distinctions, because population averages do not represent constraint relationships. They represent variant frequency.
The diabetes architecture has redundancy. The hematology architecture has less. Both are real biology. Neither is anomalous. A framework that reads each one correctly produces a reconfiguration signal in the first case and a progressive boundary signal in the second. A framework that imposes one interpretation on every cohort would produce the same shape of output regardless of underlying biology.
That is the test the field has been missing. Architectural reading is not a methodology that overlays a structure on the data. It is a framework that lets the data's actual structure express itself. Two cohorts with two different architectural realities produce two different empirical signatures under the same analytic methodology. The framework is not generating the signal. It is reading what is already there.
What this means for variant interpretation, drug discovery, and pharmacogenomic prediction.
Two patients with similar individual variant profiles can have different constraint architectures. A drug engaging variants in a redundant network produces reorganization toward an alternative coherent configuration. A drug engaging variants in a non-redundant network produces progressive constraint depletion. Same molecular targets. Different clinical outcomes. The variability is not noise. It is signal that current pharmacogenomic infrastructure cannot read.
This is the failure mode underneath much of the response heterogeneity that drug development has been hitting for years. Trials that fail in subpopulations the standard frameworks cannot stratify. Pharmacogenomic predictions that work for some patients and not for others. Clinical genomics interpretations that produce reclassifications as new context emerges. Each of these is the empirical signal of a structural problem in how the field reads biology.
Architecture-aware reading is the structural fix. It does not replace the existing ACMG/AMP framework. It extends it by adding constraint-network context, evolutionary architecture, and combinatorial function as evidence categories alongside the existing weights. The components to do this exist. The infrastructure is becoming available. The empirical evidence is now in front of us.
The next decade is built by reading architecture.
White-hat biological hacker work, in the architectural sense. The system has structure. The structure is readable. The platform reads it. The empirical signals are reproducible across runs and architecturally distinguishable across domains.
Two domains. One framework. Two distinct architectural signatures. The next decade of variant interpretation, drug discovery stratification, and pharmacogenomic prediction will be built by infrastructure that reads architecture, not by infrastructure that scores variants in isolation.
The empirical evidence is no longer theoretical. It is in front of us now.
This work has been compounding for four years and the framework is producing the kind of run-by-run signal the architecture predicted. Diabetes shows reconfiguration. Hematology shows progressive boundary. Both are real. Both are reproducible. Both demonstrate distributed constraint architecture as the empirical reality of how variant networks actually function.
The architecture is reading two diseases differently. That is the point.